Three-Dimensional Microstructure Modeling
This page summarizes earlier research work by Martin O. Steinhauser on three-dimensional microstructure modeling, polycrystalline materials, mesoscopic simulation, and the generation of realistic grain structures. The central question was how the geometry of a material’s microstructure influences its macroscopic mechanical behavior.
Polycrystalline materials consist of many grains with different orientations, shapes, and interfaces. Their effective properties depend not only on the material composition, but also on grain size, grain boundaries, defects, and spatial organization. For many engineering materials, the relation between microstructure and macroscopic response is therefore a central problem.
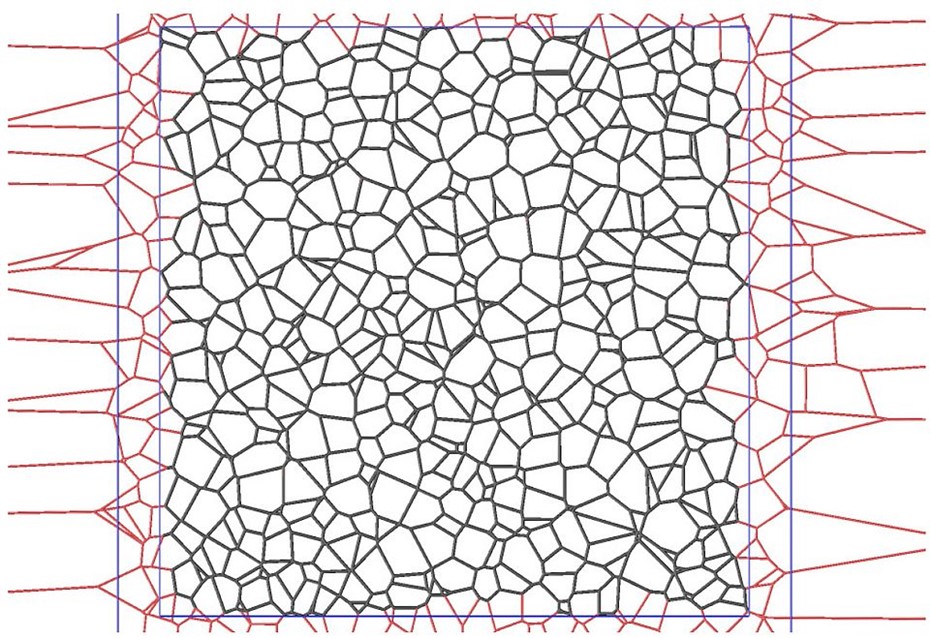
Figure 1 shows an experimental micrograph of a polycrystalline material. Such images provide information about grain shapes and grain-boundary networks, but they do not directly provide a complete three-dimensional model. Figure 2 illustrates a generated three-dimensional microstructure that represents a polycrystalline grain system. Figures 3 and 4 show further model structures used to investigate mesoscopic organization and the mechanical or statistical properties of such systems.
Concept
The concept of this research line was to construct realistic computational representations of microstructures and to use them as input for numerical analysis. Instead of treating a material as homogeneous from the outset, the microstructure itself becomes part of the model.
This approach connects experimental image information, geometric reconstruction, statistical characterization, and numerical simulation. Methods such as power diagrams, Voronoi-type constructions, discrete-element approaches, and finite-element modeling can be used to generate and analyze microstructures with controlled statistical properties.
Applications
Applications include polycrystalline materials, ceramics, hard materials, porous structures, grain-boundary networks, and materials whose macroscopic behavior is governed by microstructural details. Such models are relevant when strength, hardness, brittleness, ductility, fracture, or transport properties depend on grain geometry and interfacial structure.
The broader relevance of this earlier work lies in connecting experimentally observed microstructures with computational models. It contributes to the general problem of deriving effective material behavior from mesoscopic structure rather than assuming it phenomenologically.
The publication listed below documents the modeling approach, geometric construction methods, and numerical analysis associated with this research line.
Selected Related Publications
Modeling and Simulation of Microstructures using Power Diagrams: Proof of Concept
M. Kühn, M.O. Steinhauser
Appl. Phys. Lett. 2008, 93, 1